Clinical Studies
Principle Investigator at University Health Network, Princess Margaret Cancer Centre, Dr. Girish Kulkarni MD, Surgeon-Scientist affiliated with the Department of Surgery, Faculty of Medicine at the University of Toronto discusses the successful completion of the first human clinical study for Theralase’s Anti-Cancer Study Drug, TLD-1433.
Phase Ib NMIBC Clinical Study
The Phase Ib NMIBC clinical study (“Study I”) is entitled,
“A Phase Ib Trial of Intravesical Photodynamic Therapy in Patients with Non-Muscle Invasive Bladder Cancer at High Risk of Progression Who are Refractory to Therapy with Bacillus Calmette-Guérin (“BCG”) and Who are Medically Unfit for or Refuse a Cystectomy”.
Study I's purpose was to evaluate TLD-1433, Theralase®’s lead PDC for the primary objective of safety and tolerability, a secondary objective of pharmacokinetics (movement and exit of drug within tissue) and tertiary objective of efficacy, primarily at 90 days and secondarily at 180 days post treatment.
Study Design
Theralase® has completed a Phase Ib NMIBC clinical study (“Study I”), with 6 patients treated at University Health Network (“UHN”) (Toronto, Ontario, Canada).
- Three patients were treated with the Maximum Recommended Starting Dose (“MRSD”) of the Study Drug (TLD-1433) instilled intravesically into their bladder, light activated by fibre optics placed inside the bladder connected to the study device (TLC-3200 Medical Laser System) at a specific target energy density.
- The MRSD of the study drug was proven safe after independent review by the Data Safety Monitoring Board (“DSMB”) and an additional three patients were enrolled and treated at the therapeutic dose (2 times the MRSD) of the study drug (TLD-1433) instilled intravesically into their bladder, light activated by fibre optics placed inside the bladder connected to the study device (TLC-3200 Medical Laser System) at a specific target energy density.
Study Outcome Objectives:
1. Primary
Evaluate safety and tolerability. Primary objective of safety achieved after 6 patients treated in Study I experienced only grade 1 and grade 2 Adverse Events (“AEs”) (e.g.: bladder spasms, constipation, urge incontinence, fatigue, urinary frequency, hematuria, penile discomfort, urinary urgency, pain and other), where 95% of these AEs resolved within 180 days. For the 5% of AEs not completely resolved, all were considered pre-existing conditions in the opinion of the Principal Investigator (“PI”) and hence none were deemed related to Study I.
2. Secondary
Evaluate the pharmacokinetics. Secondary objective achieved at the therapeutic dose: 89.0% of the Study Drug was excreted from urine within 24 hours and 99.9% was excreted within 72 hours post study treatment. In plasma, 68% was removed within 24 hours and 90% removed within 72 hours post study treatment. See Figure 1.
3. Exploratory
Evaluate efficacy. Complete Response ("CR") rates were demonstrated in 67% of patients (2 out of 3 patients) treated at the therapeutic dose of the study drug at the 90 day assessment (urine cytology and cystoscopy). This CR rate of 67% (2 out of 3 patients) continued up to 450 days post initial CR (540 days post study treatment).
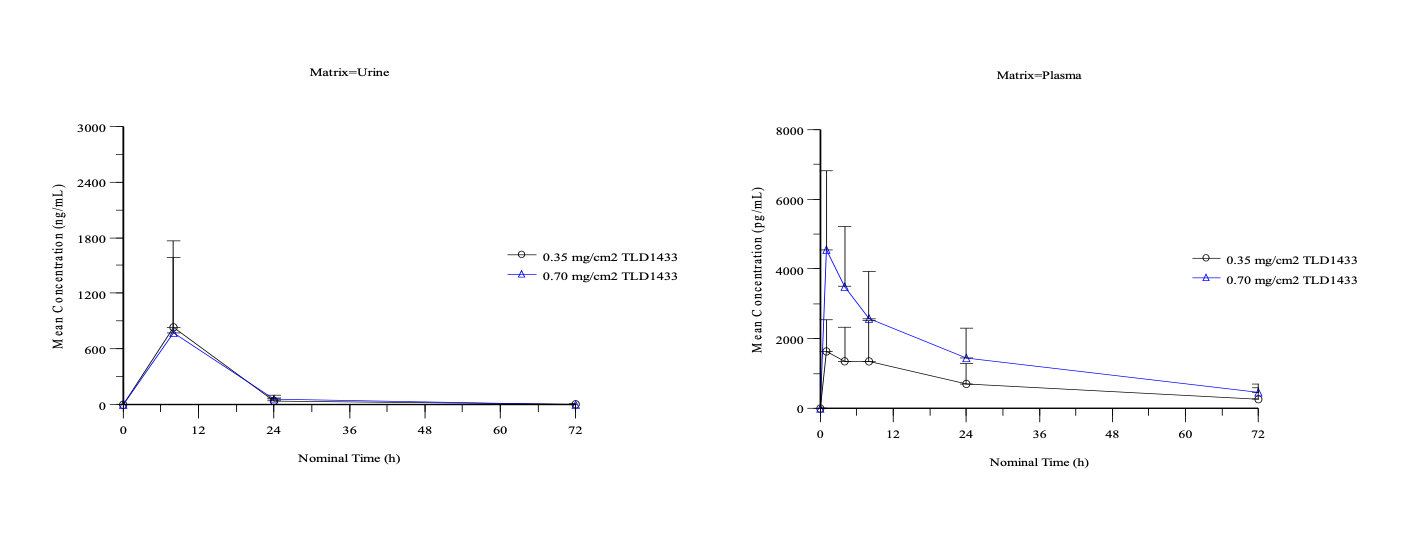
This latest data validates the strong efficacy signal that the Company has received after only a single ACT treatment in the completed Study I, representing a 67% CR in the therapeutic dose group.
The Medical and Scientific Advisory Board (“MSAB”) recommended that the clinical data collected from six patients in Study I were sufficient to support the Study I objective and conclude that Study I had successfully achieved it’s primary, secondary and tertiary objectives at the therapeutic dose and had adequately addressed the study’s scientific, technical and clinical questions, as per the approved study design and clinical protocol. The MSAB recommendation to the Company was to successfully complete Study I based on the six patients treated and suggested that the Company immediately pursue a pivotal Phase II NMIBC clinical study approval with Health Canada and the Food and Drug Administration (“FDA”) to commence Study II, with a primary objective of efficacy.
FDA guidelines (February 2018) for BCG-Unresponsive Non Muscle Invasive Bladder Cancer states that:
“In BCG-Unresponsive NMIBC, a single-arm clinical trial with Complete Response ("CR") rate and duration of response as the primary endpoint can provide primary evidence of effectiveness to support a marketing application”
For NMIBC, a CR is defined by the FDA as the definitive endpoint for single-arm intravesical studies of patients who present with BCG-Unresponsive Carcinoma In-Situ (“CIS”) disease, with or without resected papillary tumours, at any point in time and to demonstrate duration of CR at 360 days post initial CR.
Study Treatment
Theralase’s ACT involves the intravesical instillation of a water-based solution of the lead PDC, TLD-1433, via a catheter inserted through the urethra into the patient’s bladder, allowing the PDC to be preferentially absorbed by NMIBC tumours.
The bladder is then drained of the solution, flushed with sterile water to remove non-absorbed TLD-1433 and refilled with sterile water via a cystoscope.
The Study Device (TLC-3200), is a fiber optic assembly, containing a Laser Emitter used to emit laser light and a Laser Detector used to measure the intensity of laser light delivered. When the Study Device is inserted through the cystoscope and once positioned, the laser light is emitted to activate the study drug absorbed in the cancer cells to produce singlet oxygen and Reactive Oxygen Species ("ROS") to induce the cancer cell into Immunogenic Cell Death ("ICD") through oxidative stress.
Pivotal Phase II NMIBC Clinical Study
In April 2019, Theralase has commenced Phase II NMIBC Clinical Study.
The pivotal Phase II NMIBC Clinical Study, entitled:
“A Phase II Clinical Study of Intravesical Photo Dynamic Therapy in Patients with BCG-Unresponsive Non-Muscle Invasive Bladder Cancer or Patients Who are Intolerant to BCG Therapy” (“Study II”)
Study II will utilize the Therapeutic Dose (0.70 mg/cm2) of TLD-1433 and will focus on the enrollment and treatment of approximately 100 to 125 BCG-Unresponsive NMIBC patients presenting with CIS in approximately 20 clinical sites located in Canada and the United States, with a primary endpoint of efficacy, a secondary endpoint of duration of efficacy and a tertiary endpoint of safety.
IMPORTANT INFORMATION FOR PATIENTS
If you are a patient interested in participating in the Phase II NMIBC clinical study (“Study II”), please feel free to contact Theralase® or the nearest participating clinical study site for more information.
If enrolled in Study II, there is a no charge for participation. for patients who qualify; however, your urologist will need to contact the principal investigator to provide confidential information about your NMIBC to ensure you meet the clinical study guidelines.
For patients or practitioners wishing to learn more about Study II, please visit ClinicalTrials.gov and search for "Theralase" or by the National Clinical Trial Number: NCT03945162.
Medical and Scientific Advisory Board

Ashish M. Kamat
MD, MBBS, FACS, Professor of Urology, MD Anderson
Internationally recognized expert in urologic oncology and an authority in the management of urologic cancers with expertise in bladder cancer, organ sparing and minimally invasive techniques. Maintains an active research portfolio with focus on efforts to develop novel therapies and identify predictors of response to therapy. Initiated, led and active in several large studies including multinational trials in bladder cancer, findings published in high impact journals.Dr. Michael A. O’Donnell
MD, Professor of Urology, University of Iowa
Long history of focusing on bladder immunology and bladder cancer immunotherapy, particularly the anticancer mechanism of Bacillus Calmette-Guerin (“BCG”) and its enhancement with combination therapies. Recently headed a national trial of bladder cancer treatment utilizing BCG plus interferon (a natural protein which induces health cells to combat disease) comprised of over 1,000 patients and holds several US patents for his work.


Expertise in biomedical
and clinical research
Our Medical and Scientific Advisory Board ("MSAB") is comprised of Key Opinion Leaders ("KOP") with broad expertise in clinical research and development.
The MSAB members meet with the Theralase® team to review the clinical study data to provide critical evaluation and guidance on current programmes to ensure that Theralase®'s research is clinically supported.
Research Team
Unique combination
of scientific preclinical and clinical capabilities
Theralase® has a unique combination of scientific preclinical and clinical capabilities in small light-activated molecules discovery, design and development. This puts Theralase® in a strong position to develop the targeted innovative, science based, medical technology platforms required to meet patient and healthcare practitioner needs.
To preserve the value of Theralase®'s cutting-edge science and technology, the Company employs a comprehensive Intellectual Property ("IP") strategy through the filing of worldwide patents.
Arkady Mandel
MD, PH.D., D.SC, Chief Scientific Officer
More than 34 years experience as a clinical and scientific researcher of medical laser technology used to safely and effectively treat numerous medical conditions.Mark Roufaiel
Research Scientist, PhD
Immunolgy - Cardiovascular BiologyPavel Kaspler
Research Scientist, PhD



Our Partners
As part of our continuing mission to design, develop, and manufacture cutting-edge laser technologies, Theralase® has partnered with individuals and groups at the following institutions and organizations.









