Toronto, Ontario – May 30th, 2024, Theralase® Technologies Inc. (“Theralase®” or the “Company”) (TSXV: TLT) (OTCQB: TLTFF), a clinical stage pharmaceutical company dedicated to the research and development of light and/or radiation activated Photo Dynamic Compounds (“PDCs”) for the safe and effective destruction of various cancers, bacteria and viruses has released the Company’s unaudited interim consolidated 1Q2024 financial statements (“Financial Statements”).
Theralase® will be hosting a conference call on Thursday June 6th, 2024 at 11:00 am ET, which will include a presentation of the financial and operational results for the fiscal quarter ending March 31st, 2024. Questions are welcome; to ensure we have time to review and answer them during the call, please send them in advance to mperraton@theralase.com.
Zoom Meeting Link: https://us02web.zoom.us/j/89087760875
Conference Call in: 1-647-558-0588 (Canada) / 1-646-558-8656 (US) – not required for those attending by Zoom.
An archived version will be available on the website following the conference call.
Financial Summary:
For the three-month period ended March 31st:
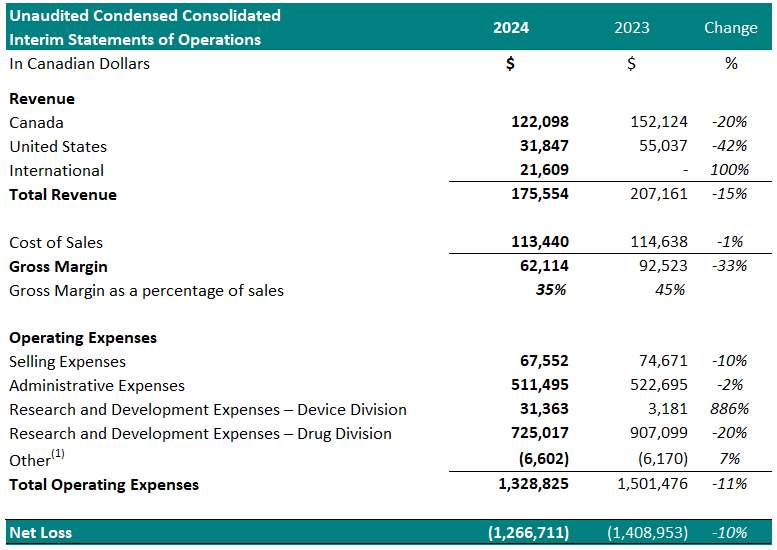
1 Other represents foreign exchange, interest accretion on lease liabilities and / or interest income
For the three-month period ended March 31st, 2024;
- Total revenue decreased 15%, year over year.
- Cost of sales was $113,440 (65% of revenue) resulting in a gross margin of $62,114 (35% of revenue). In comparison, the cost of sales for the same period in 2023 was $114,638 (55% of revenue) resulting in a gross margin of $92,523 (45% of revenue). The gross margin decrease as a percentage of sales, year over year, is attributed to an increase in material costs.
- Selling expenses decreased to $67,552, from $74,671 for the same period in 2023, a 10% decrease. The decrease is a result of reduced spending in commissions (26%), and travel (74%).
- Administrative expenses decreased to $511,495 from $522,695 for the same period in 2023, a 2% decrease. The decrease is a result of reduced spending on general and administrative expenses (43%), insurance costs (8%) and stock based compensation (10%).
- Stock based compensation expense decreased 10% in 2024, due to the cumulative effect of accounting for the vesting of stock options granted in the current and previous years.
- Net research and development expenses for the Drug Division decreased to $725,017 from $907,099 for the same period in 2023, a 20% decrease. The decrease is primarily attributed to a decrease in costs for Study II patient enrollment and treatment.
- Net research and development expenses for the Device Division increased to $31,363 from $3,181 for the same period in 2022, an 886% increase. The increase is attributed to development of a new software program for the TLC-2000 Cool Laser Therapy system.
- Net loss was $1,266,711, which included $220,919 of net non-cash expenses (i.e.: amortization, stock-based compensation expense and foreign exchange gain/loss). This compared to a net loss in 2023 of $1,408,953, a 10% year over year reduction, which included $244,787 of net non-cash expenses. The Drug Division represented $1,011,762 of this loss (80%) in 2024. The decrease in net loss is primarily attributed to decreased spending on research and development expenses in Study II.
Operational Highlights:
Non-Brokered Private Placement
On April 24, 2024, the Company closed a non-brokered private placement of units. On closing, the Company issued an aggregate of 4,167,778 units at a price of $0.18 per Unit for aggregate gross proceeds of approximately $750,200. Each Unit consists of one common share of the Company and one non-transferable common share purchase warrant. Each Warrant entitles the holder to acquire an additional Common Share at a price of $0.25 for a period of 5 years following the date of issuance.
In 2024, the Company plans to secure funding through various equity and debt instruments to allow the Company the ability to become base shelf eligible. This will allow the Company sufficient funding to complete enrollment into Study II by year end, data lock in mid 2026 and position the Company for FDA and Health Canada approval by the end of 2026, subject to achieving FDA Priority Review.”
Study II Update
On February 8th, 2024, Dr. Michael Jewett joined the Company in the role of an independent consultant, to assist the Company in the accruement of patients into Study II. Under the terms of the consulting agreement, Dr. Jewett will be responsible for working with existing clinical study sites and helping to onboard new clinical study sites to assist Theralase® to complete enrollment and provide the primary study treatment to all 100 patients in Study II, by December 31, 2024.
To date, Study II has provided the primary study treatment for 68 patients, with new patients being enrolled in 2Q2024.
Theralase® is working to add up to 5 new CSSs in 2024, as well as increase enrollment at the existing 10 CSSs to complete Study II accruement by the end of 2024.
All patients received the primary Study Procedure (n=68).
Approximately 50% of the patients received the maintenance Study Procedure (n=33)
Of the 33 patients who received the maintenance Study Procedure, 25 were CR prior to treatment, 4 were IR and 4 were No Response (“NR”) (positive cystoscopy and positive urine cytology).
In conclusion, the primary Study Procedure provides a 58% opportunity for CR at the 90 day assessment.
An advisory board meeting was completed on April 12, 2024 during the Canadian Urologic Association (“CUA”) Bladder Cancer Forum 2024 located in Toronto, Ontario and provided an update to all Canadian PIs of the Study II interim clinical data and an opportunity to discuss patient enrollment.
An advisory board meeting was completed on May 4th, 2024 during the 2024 American Urology Association (“AUA”) meeting in San Antonio, Texas and provided an update to all attendees of the Study II interim clinical data and an opportunity to discuss patient enrollment.
Break Through Designation Update
The Company submitted a pre-Break Through Designation (“BTD”) submission to the FDA in July 2023 and based on the FDA’s feedback, the Company is currently working with the Clinical Study Sites (“CSSs”), a biostatistics organization and a regulatory organization to update the pre-BTD with clinical data clarifications identified by the FDA. The Company plans to resubmit the pre-BTD submission to the FDA in 2Q2024/3Q2024 for FDA review of these clarifications. Once the pre-BTD submission has been accepted by the FDA, the Company will compile a BTD submission for review by the FDA in support of the grant of a BTD approval.
Theralase® has commenced receiving clinical data from the CSSs with a number of patients showing a duration of their CR beyond 450 days, with some patients demonstrating CR for up to 3 years, post the primary Study Procedure.
Additional clinical data is required, prior to a pre-BTD submission to the FDA.
Study II Preliminary Clinical Data:
Performance to Primary, Secondary and Tertiary Objectives
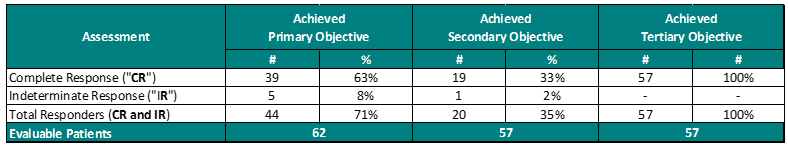
For the primary objective, 63% of patients provided the Study Procedure (Study Drug activated by the Study Device) demonstrated a Complete Response (“CR”) (negative cystoscopy and negative urine cytology). Including patients, who demonstrated an Indeterminate Response (“IR”) (negative cystoscopy and positive or suspicious urine cytology), the Total Response (“TR”) increases to 71%. This represents approximately 3 out of 4 Bacillus Calmette Guérin (“BCG”)-Unresponsive Non-Muscle Invasive Bladder Cancer (“NMIBC”) Carcinoma In-Situ (“CIS”) patients treated with Theralase®’s unique Study Procedure are demonstrating complete destruction of their CIS bladder cancer within their bladders.
For the secondary objective, 33% (approximately 1 out of 3) patients demonstrated a duration of their CR for 15 months from date of first treatment with 35% of patients demonstrating a TR.
For the tertiary objective, no patients have been diagnosed with a Serious Adverse Event (“SAE”) directly related to the Study Drug or Study Device.
Note: One patient is currently under investigation for their secondary objective performance, as they demonstrated a CR, potentially recurred and demonstrated a CR once again.
Clinical Data Based on Assessment Visit:
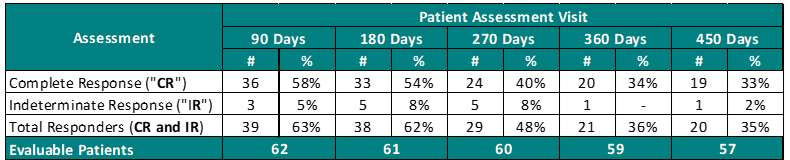
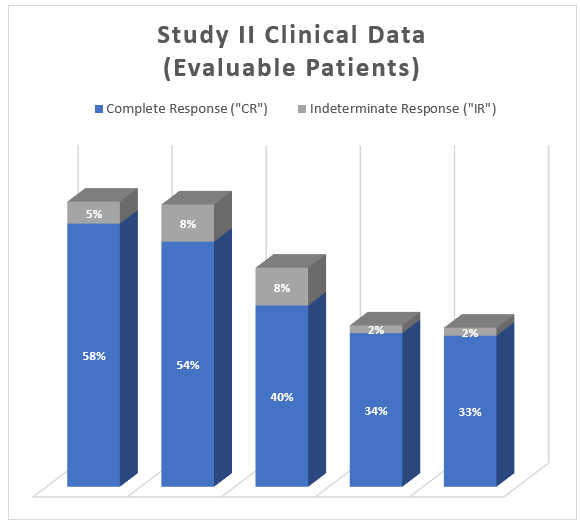
The interim clinical data demonstrates that at the 90 Day Assessment, 58% of Evaluable Patients achieved a CR and 63% achieved a Total Response (CR + IR) post primary Study II Treatment and at the 450 Day Assessment 33% achieved a CR and 35% achieved a TR.
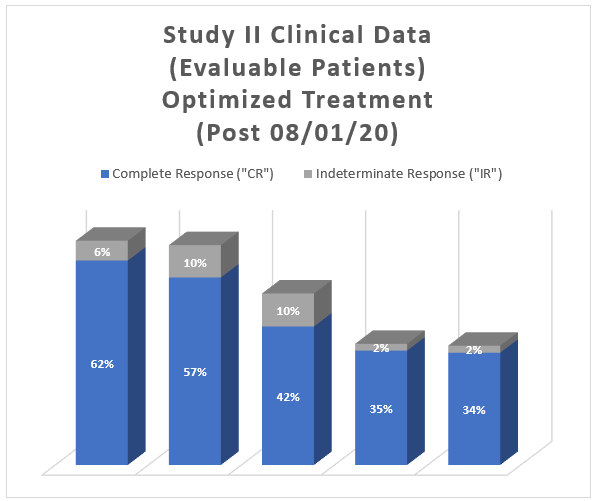
Study II Clinical Data Based on Assessment Visit for Patients Treated with the Optimized Study Procedure (Post August 1, 2020):
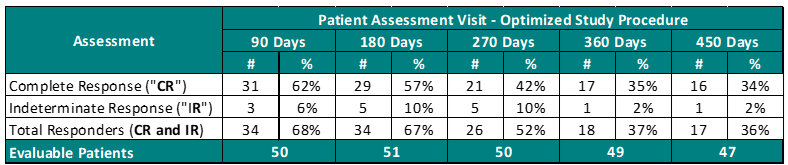
The above interim clinical data demonstrates that for patients who received the Optimized Study Procedure at the 90 Day Assessment visit, 62% of Evaluable Patients achieved a CR and 68% achieved a Total Response (CR + IR) post primary Study Procedure. At the 450 Days Assessment, 34% achieved a CR and 36% achieved a TR.
Note:
Patient Response Chart:
The Swimmer’s plot below is a graphical representation of the interim clinical results (n=68) graphically demonstrating a patient’s response to a treatment over time. As can be seen in the plot, clinical data is still pending for patients, who have demonstrated a CR and continue to demonstrate a duration of that response.
Swimmer’s Plot:
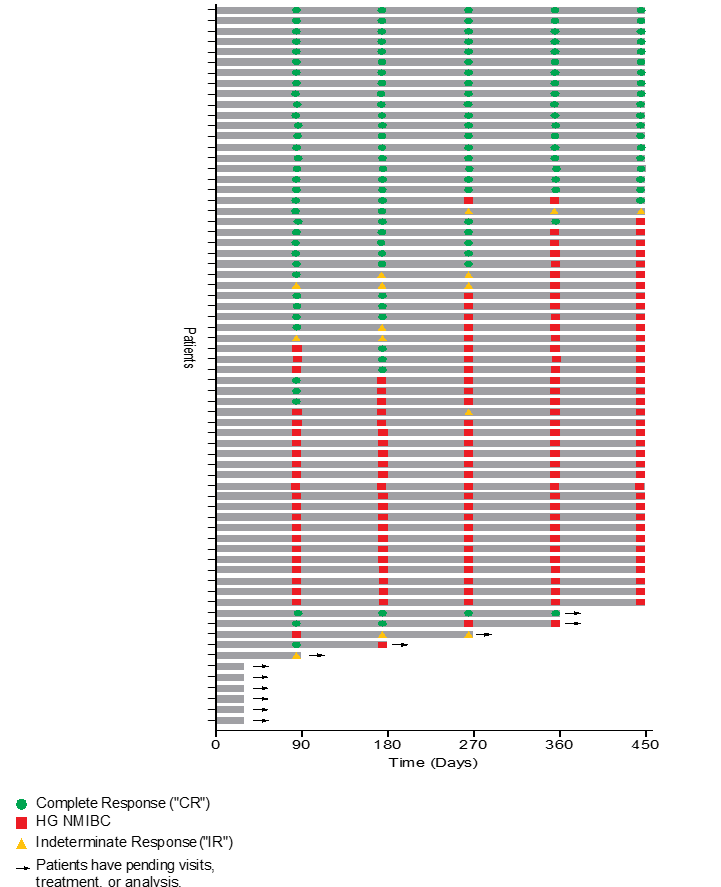
The Swimmer’s Plot illustrates:
- 19 Evaluable Patients achieved CR initially and at the 450 days assessment and thus achieved the primary and secondary objectives of Study II for all patients assessed up to 450 days (19/57 = 33%).
- 39 Evaluable Patients that achieved CR on at least one assessment date and thus achieved the primary objective of Study II (39/62 = 63%)
Kaplan-Meier Curve
The Kaplan-Meier (“KM”) Curve represents the interim cumulative incidence of clinical events, including the treatment efficacy, occurring over prespecified time in Study II.
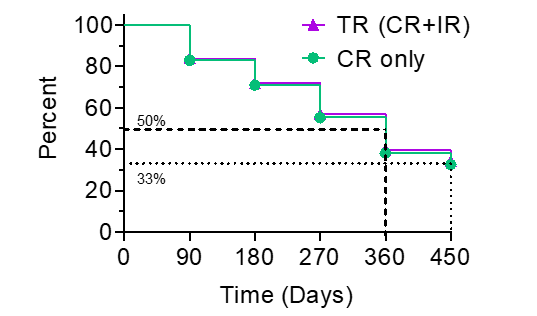
According to the interim clinical data in the KM curve:
- > 80% of patients remained in Study II after 90 days, following the initial Study Procedure.
- 35% of Total Response patients have a duration of response ? 450 days.
- 33% of Complete Response patients have a duration of response ? 450 days.
Serious Adverse Events
For 68 patients treated in Study II, there have been 13 Serious Adverse Events (“SAEs”) reported:
- 2 – Grade 2 (resolved within 1 and 1 days, respectively)
- 7 – Grade 3 (resolved within 1, 2, 3, 4, 4, 82 and unknown days, respectively)
- 3 – Grade 4 (resolved within 3, 6 and 8 days, respectively)
- 1 – Grade 5
Theralase® believes all SAEs reported to date are unrelated to the Study II Drug or Study II Device, as reviewed and confirmed by an independent Data Safety Monitoring Board (“DSMB”).
Note: A SAE is defined as any untoward medical occurrence that at any dose: Is serious or life-threatening, requires inpatient hospitalization or prolongation of existing hospitalization, results in persistent or significant disability/incapacity, is a congenital anomaly/birth defect or results in death.
Dr. Arkady Mandel, M.D., Ph.D., D.Sc., Chief Scientific Officer of Theralase® stated, “The interim clinical data of Study II to date has proven to be world-class. Study II has demonstrated an ability to destroy urothelial cell carcinoma in a patient’s bladder for a total response of 71% and a duration of that total response of 36%, for patients treated with the optimized Study Procedure. The primary benefits of the Theralase® technology versus competitive technologies are the: urologist-led treatment, single out-patient treatment, high efficacy rates (patients achieve a CR in 58% of the cases after only one Study Procedure), high duration of response (up to 3 years) and high safety margin (no SAEs directly associated with the Study Drug or Study Device); therefore, the Theralase® technology presents a safe, effective alternative therapy for patients, who are at risk of having their bladder removed.”
About Study II:
Study II utilizes the therapeutic dose of the patented Study II Drug ( “RuvidarTM” or “TLD-1433”) (0.70 mg/cm2) activated by the proprietary Study II Device (TLC-3200 Medical Laser System or “TLC-3200”). Study II is focused on enrolling and treating approximately 100 BCG-Unresponsive NMIBC Carcinoma In-Situ (“CIS”) patients in up to 15 Clinical Study Sites (“CSS”) located in Canada and the United States.
About RuvidarTM:
RuvidarTM is a peer reviewed, patented PDC currently under investigation in Study II.
About Theralase® Technologies Inc.:
Theralase® is a clinical stage pharmaceutical company dedicated to the research and development of light activated compounds, their associated drug formulations and the light and/or radiation systems that activate them, with a primary objective of efficacy and a secondary objective of safety in the destruction of various cancers, bacteria and viruses.
Additional information is available at www.theralase.com and www.sedarplus.ca
Neither TSX Venture Exchange nor its Regulation Services Provider (as that term is defined in the policies of the TSX Venture Exchange) accepts responsibility for the adequacy or accuracy of this release.
Forward Looking Statements:
This news release contains “forward-looking statements” within the meaning of applicable Canadian securities laws. Such statements include; but, are not limited to statements regarding the Company’s proposed development plans with respect to Photo Dynamic Compounds and their drug formulations. Forward looking statements may be identified by the use of the words “may, “should“, “will“, “anticipates“, “believes“, “plans“, “expects“, “estimate“, “potential for” and similar expressions; including, statements related to the current expectations of Company’s management for future research, development and commercialization of the Company’s Photo Dynamic Compounds and their drug formulations, preclinical research, clinical studies and regulatory approvals.
These statements involve significant risks, uncertainties and assumptions; including, the ability of the Company to: adequately fund and secure the requisite regulatory approvals to commercially market a treatment for bladder cancer in a timely fashion and implement its commercialization strategy. Other risks include: the ability of the Company to successfully complete its Phase II BCG-Unresponsive NMIBC CIS clinical study , access to sufficient capital to fund the Company’s operations may not be available or may not be available on terms that are commercially favorable to the Company, the Company’s drug formulations may not be effective against the diseases tested in its clinical studies, the Company’s fails to comply with the term of license agreements with third parties and as a result loses the right to use key intellectual property in its business, the Company’s ability to protect its intellectual property, the timing and success of submission, acceptance and approval of regulatory filings. Many of these factors that will determine actual results are beyond the Company’s ability to control or predict.
Readers should not unduly rely on these forward- looking statements, which are not a guarantee of future performance. There can be no assurance that forward looking statements will prove to be accurate as such forward looking statements involve known and unknown risks, uncertainties and other factors which may cause actual results or future events to differ materially from the forward-looking statements.
Although the forward-looking statements contained in the press release are based upon what management currently believes to be reasonable assumptions, the Company cannot assure prospective investors that actual results, performance or achievements will be consistent with these forward-looking statements.
All forward-looking statements are made as of the date hereof and are subject to change. Except as required by law, the Company assumes no obligation to update such statements.
For investor information on the Company, please feel to reach out Investor Inquiries – Theralase Technologies.
For More Information:
1.866.THE.LASE (843-5273)
416.699.LASE (5273)
www.theralase.com
Kristina Hachey, CPA
Chief Financial Officer
khachey@theralase.com